


CRS4 talks to Aaron Ciechanover, 2004 Nobel Prize winner for Chemistry, about personalized medicine and innovative cancer therapies1
Bortezomib! Who would that be?
Is it the name of a Babylonian king? A three litre bottle of champagne? The index of the technology sector on the Bordeaux stock exchange? The answer is, obviously, none of these. Bortezomib, which has been available for a few years, is the forerunner of a family – which we hope is destined to grow – of new-generation drugs for the treatment of multiple myeloma.
As I prepare to meet2 Aaron Ciechanover, who won the 2004 Nobel Prize for Chemistry with Irwin Rose and Avram Hershko, I reflect on the innovative therapies offered by personalized medicine, on the applications of molecular biology in clinical oncology and on the expectations of so many cancer patients.

Their research led to the discovery of the ubiquitin system (as usual, we redirect our more curious readers to the Technical corner). The story unwinds across years and in laboratories on different continents, with a fascinating mix of methodological effort, creativity, chance, mistakes, successes, and human relations. Bortezomib, the active ingredient that is based on the inhibition of proteasome, is in fact the first significant therapeutic application directly linked to the ubiquitin system and the regulation thereof. And so we thought that Aaron Ciechanover would be the perfect person to speak to about innovative cancer drugs and personalized medicine.
Professor Ciechanover, we heard that Irwin “Ernie” Rose, who won the Nobel Prize for Chemistry with you and Avram Hershko in 2004, passed away yesterday. We know that he meant a lot to you, as a colleague and as a friend, and we hope that you will accept our condolences. Irwin Rose was much older than you and your relationship was initially one of student and professor?
Thank you. Yes, in truth my professor [Avram Hershko3] and I were both guests in Irwin Rose’s laboratory. I was 10 years younger than my professor who in turn was 10 years younger than Irwin.
About ten years ago, the first sequencing of a human genome required approximately 3 billion US dollars and 13 years of analyses. Today the same result can be obtained at a cost of a few thousand US dollars (depending on the coverage) in just a few days: one expects that the cost will diminish even further and that it will become possible to carry out the analyses in 24 hours. This has opened the doors to “personalized medicine”: in your opinion, what is the current status of this discipline, and what developments can we expect in the immediate future?
I think that DNA sequencing will become a routine test, like X-rays or magnetic resonance. DNA will only be the start: there are other more complicated aspects that we have to keep in mind, such as RNA, proteins, metabolites, etc. Fortunately there are certain principles and everyday technologies that will facilitate these analyses. I believe that we will eventually be able to obtain a complete molecular profile for the treatment of sick people of our day and in the future.
The Italian National Health Service has adopted a “pay-by-result” system for the calculation of drug prices, thanks to which the average cost of treatment is among the lowest in Europe. Even so, the cost of cancer drugs has more than doubled in the last 10 years; it is considered normal for the haematology ward in a hospital to spend one third of its budget for purchasing medicines. This problem is not limited to oncology: for example, many people today are upset about the cost of new treatments for Hepatitis C. Why do new generation drugs have such astonishingly high prices?
It is difficult to say, because I am not familiar with the economic inner workings of the pharmaceutical industry. In actual fact, new medicines could be more expensive. The cost of a new drug that is released onto the market is in the order of billions of US dollars; what is more, many experimental drugs fail phase III of the clinical trials, giving rise to expenditure that will never be recovered. Then let’s not forget that often the big pharmaceutical houses have to face serious legal claims for compensation because of possible damages caused by the side effects of the drugs, for example cardiotoxicity. And so you see that the development of new drugs costs a lot, even those that will never reach the market, and many drugs have got a very short life, for example antibiotics.
The advent of personalized medicine could pose a serious problem for pharmaceutical giants. For example, there are a limited number of drugs (statins) currently available to control cholesterol levels: they work well for some people, not so well for others, in still others they cause side effects. But everyone uses them because there are no noteworthy alternatives. With personalized medicine, the number of patients would remain more or less the same, but there would be more drugs, and each one would cover only a small market segment. Similarly, we are coming to realize that there are not only two or three types of breast cancer, but many more: so the number of drugs will increase and each one will be used for a much more specific fraction of the total number of patients.
This is why I believe that the cost of new drugs is destined to increase. We will probably have to start making more use of private insurance policies, because the public health services will not be able to cover all of the costs tied to the new treatments. Not to mention the diseases that go hand in hand with the ageing of the population, such as Alzheimer’s or Parkinson’s disease.
Your discovery made the development of bortezomib possible, a proteasome inhibitor that has been successfully used for the treatment of multiple myeloma. You obtained your first results at the beginning of the 80s, while bortezomib was synthesized in 1995 and then approved by the American FDA in 20034 . Why does so much time pass between the original scientific intuition and the development of a new treatment? Is there a way to speed up this process of “research translation”?
Maybe it is possible to speed it up but, I fear, not notably. Scientific intuition, the development of a new drug and its availability for use are different matters altogether. The discovery of proteasome did not happen immediately, but it was a gradual process. It’s like making a river from a small spring, the river being the scientific credibility that you have to earn in the eyes of your colleagues: for example, it was thought that inhibiting proteasome would have irreparable consequences, and so nobody believed that it could be possible to use proteasome as a therapeutic target. Then certain discoveries, such as the one that was the basis of bortezomib, arrive by chance and not as the result of careful planning. In the end, I’m afraid that waiting 15-20 years between the initial concept and the availability of a drug is physiological.
There are also technological considerations, let’s take for example microRNA: we have known about it for more than 20 years, but there are still no microRNA-based drugs. In 2006 Fire and Mello won the Nobel Prize for medicine for their results in the field of RNA interference: there are still no therapeutic applications. And the list goes on: today we hear a lot of talk about CRISPRs, their potential but also the risks that they entail.
Then there are not only the scientific aspects, but also the ethical aspects. Can we “correct” genes? There is the question of eugenics: we all want tall, blonde, intelligent children... there is a lot of debate in society about these and other topics: this is a possible factor that delays scientific research, and it is rightly so. It is better for society to be reasonably slow (which doesn’t mean taking centuries) but with the certainty that the new therapeutic molecule will strike the target for which it has been designed with precision and selectivity.
On the other hand there are the expectations of patients for whom no effective treatment exists, who rely on scientific research to make new drugs available in a relatively short time.
Without a doubt, but significant progress has been made in cancer treatment. Let’s take, for example, the use of T-cell modulation in the treatment of melanoma5. The clinical trials can take a lot of time, around 5 years or more, and they are necessary in order to establish the real effectiveness of the new drugs and to allow for their approval: we have to be sure that the new drug is really better than what is already available. At the end of the day, unfortunately, the animals that are subjected to laboratory tests are not rabbits or mice but human beings.
The heterogeneous nature of the genetic profiles of tumour cells6 is one of the main aspects that make the fight against cancer so difficult...
Yes, in fact patients with very similar clinical pictures can have completely different genetic landscapes, so much so that they can respond very differently to the same therapy. Then there is also intratumour heterogeneity, for example between the cells of the primary tumour and those of the metastasis in the same patient. In these cases, oncologists opt for cures based on the use of different drugs, each attacking a specific target.
So the existence of a common molecular substrate in tumour cells could facilitate research for a cure that works on a wider range of pathologies. Could the ubiquitin system be the common target for the development of more comprehensive treatments?
Maybe, but only up to a certain point. For example, we know that proteasome inhibitors work for multiple myeloma and for non-Hodgkin lymphoma, but we can’t say the same for other types of tumours7. The truth is that the term “cancer” is used to define hundreds of different pathologies.
In 1984 you went back to Technion (The Israel Institute of Technology) where, in addition to a period that was scientifically stimulating, you experienced some problems: the Technion is essentially a school for engineers, who often consider the life sciences irrelevant and who went as far as to propose the closure of your laboratory. Have things changed since then?
I think so, for the better. We are starting to understand that biomedicine and the life sciences cannot survive without engineering: just think of magnetic resonance (MRI), cardiac valves, artificial organs. Today we are more aware of the importance of multidisciplinary collaboration: we don’t have a choice, specialists have to coexist and collaborate, especially in the field of medicine which faces very complex challenges. So yes, the situation has improved: it is still not perfect [smiles] but it is definitely better than it was in the past.
Something that is not easy for physicists or engineers who are used to a mechanistic/deterministic approach to understand is that sometimes molecular biomarkers for a particular pathology are based on lists of genes that are completely different, for example the commercial prognostic and predictive tools for breast cancer: it almost seems as if there is no unique “cause and effect” relationship between genetics and disease (and response to treatment)...
In psychiatry and autism the situation is even more complicated. This is a general problem, which arises from the multigenic origin of many diseases and the different contributions made by single genes to molecular pathogenesis. We need to sequence a huge number of patient genomes in order to better understand the complex network of relationships between the different genes which contribute to a disease, and then attempt to identify among them a few essential genes (if they exist) which are found at the most important “intersections” of these networks [think also of the growing importance attributed to gene pathways8].
There is a lot of debate around these matters. Some “gurus” in cancer research, such as Weinberg and Hanahan, recommend that we try to assess the problem as a whole, taking an overall view, rather than analyzing the finer details. More than a decade has passed since the first human genome was sequenced but the therapeutic repercussions are few. We also have to consider the financial aspects, because large scale studies based on new genetic profiling techniques can be very expensive. Then there is also the question of methodology, for example which approach will be most profitable for research. These are some of the many aspects which will make the sector of personalized medicine extremely complex in the coming years, and it is for this reason that we must try to view the problem as a whole rather than focus on the details.
Today more than ever medicine makes extensive use of statistics, sometimes using terminology and concepts that are difficult for patients to understand...
Statistics obviously plays a very important role, but it must be a background role in the context of personalized medicine. If a woman hears from her oncologist that she has breast cancer, and that she has, let’s say, a 60% chance of recovery, this figure derives from the oncologist’s cultural formation, his studies, his experience, what he has read in scientific literature. But in the end the patient has feelings, a life, relationships, maybe a family, children... the patient is a person, not a number.
Statistics can help to condense knowledge about a large number of patients studied in the past in order to place this patient in a particular category and to understand which treatment is best for her. This is all that we can do today, but in the future doctors will be able to make much more specific prognoses, customized for each patient. It is no coincidence that President Obama spoke about precision, adding a fifth “p” to the features of the world of medicine in the future9.
It will be a more people-oriented and less statistics-oriented world, and this is saying a lot. It will be a massive change.
— F. Maggio
Technical corner
Between 1976 and 1981, Aaron Ciechanover was carrying on his graduate studies with Avram Hershko and Irwin Rose. One of the intuitions that inspired their work was the belief that proteins synthesis and degradation are equally important for cells fitness10.
Disregarding the mainstream attitude of the time, they decided to focus on mechanisms of protein destruction. Contrary to the common belief of single enzyme degradation, they found that several components were called into play: today we believe that the ubiquitin proteasome system (UPS) consists of more than 1000 components. Quite soon they discovered an unusual heat-stable protein involved in protein degradation processes which they called APF-1 (ATP-dependent Proteolysis Factor-1). Under certain conditions, it was capable of surprisingly increasing its molecular weight: it was soon evident that such variation took place when APF-1 attached to substrate molecules in a reversible way (dissociation to substrate could happen as well).
They speculated that covalent attachment of APF-1 was a sort of “tag” with the effect to render the target substrate susceptible to selective degradation by an ATP-dependent protease, followed by the release of free APF-111.
Let's now take a step into the past. In 1975, Gideon Goldstein and colleagues first isolated a small polypeptide hormone, the ubiquitin, apparently present in both prokaryotes and eukaryotes (this is the reason for its name). Later studies revealed that, actually, evidence of presence of ubiquitin in bacteria was a mistake due to laboratory procedure error: still, the name was maintained. More importantly, in 1980, it turned out that APF-1 and ubuquitin were the same protein12.
Some 35 years later, the mechanism by which a polyubiquitinated protein is targeted to the proteasome is not completely clear, yet. We know that coordinated reactions of three different enzymes, E1s, E2s, and E3s, take place, as depicted in Figure 1. Three main steps have been identified13:
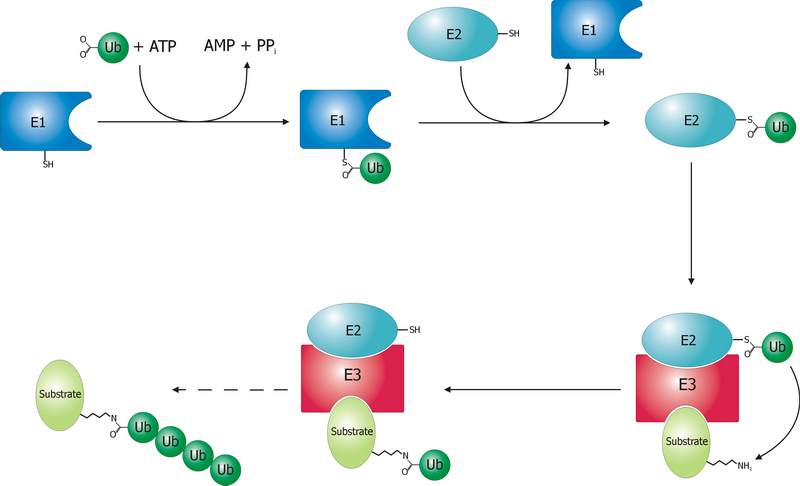
- Activation is a two-step reaction by an E1 enzyme, dependent on ATP, whose results are a ubiquitin-adenylate intermediate and the binding of ubiquitin to a cysteine residue of E1 with release of AMP. In humans two genes, UBA1 and UBA6, are associated to enzymes driving ubiquitin activation.
- Conjugation: the E2 binds to both activated ubiquitin and the E1 enzyme. There are 35 different E2 enzymes in humans, all with highly conserved structure, the UBC (ubiquitin-conjugating catalytic) fold.
- During ligation, the final step, an isopeptide bond between a lysine of the target protein and ubiquitin is created. The target protein is now “tagged” and ready for selective degradation. The human genome is supposed to contain more than 600 E3 ligases.
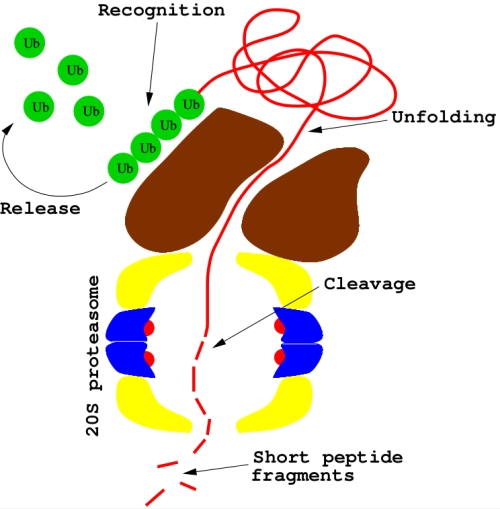
Protein degradation is then performed by the 26S proteasome, a ubiquitin-dependent large complex of proteins in the cytoplasm (see Figure 2). Substrate proteins tagged with polymeric chain of four ubiquitins or more may bind to 26S: after deubiquitination, proteins are unfolded (by the 19S subcomplex) and then delivered to the 20S proteasome, a second subcomplex where proteolysis occurrs.
Protein degradation by the UPS plays a central role in cell-cycle progression and apoptosis, DNA repair, immune response, cardiac homeostasis, response to cellular stress, et cetera. In addition, involvement of ubiquitin in a number of non-proteolytic processes as biogenesis of organelles and ribosomes, virus infection, regulation of histone modification, has emerged in recent years14.
Not surprisingly, deregulation of the ubiquitin-proteasome system may lead to human pathogenesis. In particular, since the ubiquitin-proteasome system contributes to the proper turnover of both oncoproteins and tumor suppressor proteins, aberrancies in the UPS pathway may result in malignant transformation of cells15. Thus the UPS provides a potential target for anticancer drugs.
Bortezomib main molecular mechanism is the inhibition of the 20S proteasome (see Figure 2). But why should this be a successful anticancer strategy? This is a difficult question, as many - sometimes competing - molecular processes are called into play16. As a matter of fact, cells with lower proteasome levels are significantly more vulnerable to a proteasome inhibitor, while healthy cells are less sensitive. Proteasome inhibitors may block important tumorigenic pathways, contributing also to sensitize tumors to other anticancer drugs. Furthermore, dysregulation of cell cycle can make cancer cells more susceptible to most proapoptotic stimuli, like p53. The proapoptotic protein NOXA has been recently identified as a key-agent in bortezomib-mediated toxicity in most cancer cell lines. Biological pathways somehow altered by bortezomib are actually numerous and difficult to elucidate: the hypothesis that this drug may activate different mechanisms in different patients has also been proposed.
Bortezomib comes along with two well-known limits.
First, while it is highly effective for hematological cancers, this is not the case for solid tumors. Such behaviour is quite likely explained by the low therapeutic index17 of bortezomib, whose dose-limiting toxicity is associated with peripheral neuropathy. As a consequence, proteasomes abundance in human cells is such that they greatly exceed the number of molecules of the proteasome inhibitor which can be administered to a patient18.
Inherent or acquired resistance is another major problem. In human leukemia THP1 cell-based studies, clones up to 500-fold more resistant to the drug compared with the parental cell line have been found: in that case, the \(\beta 5\) subunit (PSMB5) was overexpressed in resistant cells, resulting in a mutation in the binding site for bortezomib19. In addition, resistance can be the consequence of factors downstream of the proteasome enzymatic complex, like the overexpression of heat shock protein 27 for lymphoma cells.
To overcome these limits, an effort is being made to develop new generation proteasome inhibitors based on mechanisms different from that of bortezomib: most of them are currently in phase I/II clinical trials.
Homage to Alessandro Manzoni (Milan, 7 March 1785 – Milan, 22 May 1873)
For those who are curious to know the reason behind the title of this interview, please refer to a video-clip from the interview ↩
This interview took place on 05/06/2015 together with Andrea Mameli at the Hotel Panorama in Cagliari, whom we thank for their hospitality. A sincere thanks also goes to Saverio Gaeta and to the organisers of the Leggendo Metropolitano festival who made this meeting possible ↩
A. Ciechanover, Early work on the ubiquitin proteasome system, an interview with Aaron Ciechanover, Cell Death and Differentiation, 12, 1167–1177, 2005 ↩
Bortezomib was approved by the FDA in 2003 as a treatment for multiple myeloma in refractory patients, in 2005 as a second-line therapy, in 2008 as first-line therapy ↩
For a short review of the topic see for instance Naidoo J, Page DB, and Wolchok JD, Immune modulation for cancer therapy, BJC, 111, 12:2214-2219, 2014 ↩
Gerlinger M, Rowan AJ, Horswell S, Larkin J et al, Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing, NEJM, 366, 10:883-892, 2012 ↩
Especially solid tumors. See the Technical corner ↩
Vogelstein B, Kinzler KW, Cancer genes and the pathways they control, Nature Medicine, 10, 8:789-799, 2004 ↩
After personalized, predictive, preventive, and participatory: this is “P4 medicine”, a very popular term coined by Leroy Hood in 2005 ↩
Protein degradation is a fundamental task for eukaryotes. Inside cells, proteins homeostasis results from the opposite actions of synthesis and degradation. Proteins typical half-lives vary widely, from minutes to several days: differential rates of protein degradation is a basis of cell regulation. Regulatory molecules, such as transcription factors, are rapidly degraded, as their rapid turnover allows prompt changes to external stimuli. Degradation of other proteins in response to specific signals is another well-known mechanism for regulating the intracellular enzyme activity. Another necessary task is the destruction of faulty or damaged proteins, in order to eliminate the consequences of errors made during protein synthesis. In eukaryotes two major pathways are involved in protein degradation: the ubiquitin-proteasome system and the lysosomal proteolysis. See, for instance, Cooper GM, The Cell: A Molecular Approach. 2nd edition, Sunderland (MA) Sinauer Associates, 2000. ↩
Ciechanover A, Elias S, Heller H, Ferber S, and Hershko A, Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes, J. Biol. Chem., 255, 16:7525-7528, 1980 ↩
Wilkinson KD, Urban MK, and Haas AL, Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes, J. Biol. Chem., 255, 16:7529-7532, 1980 ↩
Hershko A, and Ciechanover A, The ubiquitin system, Annu. Rev. Biochem., 67:425–79, 1998 ↩
See Nath D and Shadan S, The ubiquitin system, Nature Insight, 458, 7237:421-467, 2009, and references therein ↩
See Table 1 in Micel LN, Tentler JJ, Smith PG, and Eckhardt SG, Role of Ubiquitin Ligases and the Proteasome in Oncogenesis: Novel Targets for Anticancer Therapies, J Clin Oncol, 31, 9:1231-1238, 2013 ↩
For a detailed analysis of molecular mechanisms of bortezomib see Cvek B, and Dvorak Z, The ubiquitin-proteasome system (UPS) and the mechanism of action of bortezomib, Curr Pharm Des, 17:1483-99, 2011 ↩
Roughly speaking, the ratio of the dose of drug that causes adverse effects divided by the dose that leads to the desired pharmacological effect ↩
Dick LR, and Fleming PE, Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy, Drug Discovery Today, 15, 5/6:243-249, 2010 ↩
Ruschak AM, Slassi M, Kay LE, and Schimmer AD, Novel Proteasome Inhibitors to Overcome Bortezomib Resistance, JNCI J Natl Cancer Inst, 103, 13:1007-1017, 2011 ↩
