


Il CRS4 parla di medicina personalizzata e terapie anticancro innovative con Aaron Ciechanover, Premio Nobel per la chimica 20041
Bortezomib! Chi era costui?
È il nome di un sovrano babilonese? La denominazione di una bottiglia di champagne da tre litri? L’indice dei titoli tecnologici della borsa di Bordeaux? Niente di tutto questo, ovviamente. Bortezomib, disponibile da pochi anni, è il precursore di una famiglia - si spera destinata a crescere - di farmaci di nuova generazione per la cura del mieloma multiplo. E proprio sulle terapie innovative promesse dalla medicina personalizzata, sulle applicazioni della biologia molecolare all'oncologia clinica e sulle aspettative dei tanti malati rifletto mentre mi accingo ad incontrare2 Aaron Ciechanover, premio Nobel per la chimica nel 2004 assieme a Irwin Rose e Avram Hershko.

Il loro lavoro di ricerca ha portato alla scoperta dell’ubiquitin system (al solito, rimandiamo i nostri lettori più curiosi al Technical corner). È una storia che si snoda attraverso gli anni e i laboratori di diversi continenti, con un mix affascinante di sforzo metodologico, creatività, serendipity, errori, successi, relazioni umane. Il bortezomib, principio attivo basato sull'inibizione del proteasoma, è di fatto la prima, significativa applicazione terapeutica direttamente collegata all’ubiquitin system e alla sua regolazione. Aaron Ciechanover ci sembra dunque l'interlocutore ideale per discutere di farmaci oncologici innovativi e medicina personalizzata.
Professor Ciechanover, abbiamo saputo che ieri è mancato Irwin “Ernie” Rose, che nel 2004 fu insignito del premio Nobel della chimica assieme a lei e Avram Hershko. Sappiamo che era una persona speciale per lei, un collega e un amico, e la preghiamo di accettare le nostre condoglianze. Irwin Rose era molto più anziano di lei, all’inizio il vostro rapporto era quello tra studente e professore?
Grazie. Sì, in realtà io e il mio professore [Avram Hershko3] eravamo ospiti entrambi nel laboratorio di Irwin Rose. C’era una differenza di 10 anni con il mio professore che a sua volta era 10 anni più giovane di Irwin.
Una trentina di anni fa, il primo sequenziamento di un genoma umano richiese circa 3 miliardi di dollari USA e 13 anni di analisi. Oggi lo stesso risultato è ottenibile ad un costo dell’ordine delle migliaia di dollari USA (variabile con il coverage) in pochi giorni di analisi: le aspettative sono che i prezzi diminuiscano ulteriormente e che l'analisi si possa effettuare in giornata. Questo ha aperto la strada alla “medicina personalizzata”: qual è lo stato attuale di questa disciplina, secondo lei, e che sviluppi avrà nel futuro immediato?
Credo che il sequencing del DNA diventerà un’esame di routine, come i raggi X o la risonanza magnetica. Il DNA sarà solo l’inizio: ci sono altre cose, più complicate, che dovremo tenere in considerazione, come RNA, proteine, metaboliti, etc. Fortunatamente ci sono alcuni principi e alcune tecnologie comuni che faciliteranno queste analisi. Alla fine credo che saremo capaci di ottenere un profilo molecolare esaustivo, per la cura delle malattie di oggi e anche di quelle future.
In Italia il Servizio Sanitario Nazionale ha adottato un criterio “pay-by-result” per la valutazione dei prezzi dei farmaci, grazie al quale il costo medio dei trattamenti è fra i più bassi d’Europa. Anche così, però, il prezzo dei farmaci oncologici è più che raddoppiato negli ultimi 10 anni; per un reparto ospedaliero di ematologia è normale spendere 1/3 del proprio budget per l’acquisto di medicine. Non è un problema esclusivo dell’oncologia: ad esempio oggi diverse persone sono irritate per il costo delle nuove terapie contro l’epatite C. Perché i farmaci di nuova generazione hanno prezzi così sorprendentemente alti?
È difficile dirlo, perché non sono addentro ai meccanismi economici delle pharma industries. In realtà le nuove medicine potrebbero essere anche più costose. La spesa tipica per un nuovo farmaco che viene immesso sul mercato è dell'ordine dei miliardi di dollari USA; inoltre molti farmaci sperimentali falliscono la fase III dei clinical trials, originando una spesa che non verrà mai ripagata. Ancora, spesso le big pharma devono affrontare in tribunale richieste pesanti di risarcimento per possibili danni causati da effetti collaterali delle medicine, come ad esempio la cardiotossicità. Così, lo sviluppo di new drugs costa molto, anche di quelle che non raggiungeranno mai il mercato, e molti farmaci hanno una vita breve, si pensi ad esempio agli antibiotici.
L'avvento della medicina personalizzata può sollevare un problema serio per le big pharma. Ad esempio, per il controllo del livello del colesterolo esiste oggi un numero limitato di farmaci (le statine): per alcuni funzionano bene, per altri meno, in alcuni causano l'insorgenza di effetti collaterali: ma tutti le prendono, perché non ci sono grandi alternative. Con la medicina personalizzata, il numero dei pazienti rimarrà più o meno lo stesso, ma i farmaci aumenteranno, e ognuno coprirà solo una fetta ristretta del mercato. Analogamente, ci stiamo rendendo conto che non esistono solo 2 o 3 tipi di tumore al seno, ma molti di più: parallelamente crescerà il numero di farmaci per questa malattia e ognuno coprirà una frazione sempre più specifica dei pazienti totali.
Per questo motivo mi aspetto che il costo dei nuovi farmaci sia destinato a salire ulteriormente. Si dovrà probabilmente ricorrere maggiormente ad assicurazioni private, perché i servizi sanitari pubblici non saranno in grado di coprire tutti i costi legati alle nuove terapie. Per non parlare delle patologie legate all'invecchiamento della popolazione, come il morbo di Alzheimer o la malattia di Parkinson.
La vostra scoperta ha permesso di sviluppare il bortezomib, un inibitore del proteasoma utilizzato con successo nel trattamento del mieloma multiplo. I vostri primi risultati risalgono agli inizi degli anni ’80, mentre il bortezomib è stato sintetizzato nel 1995 e infine approvato dalla FDA degli USA nel 20034. Perché passa così tanto tempo tra l’intuizione scientifica originale e lo sviluppo di una nuova terapia? C’è un modo di rendere questo processo di “research translation” più veloce?
Forse è possibile accorciare questi tempi ma, temo, non in modo eclatante. L’intuizione scientifica, lo sviluppo di un nuovo farmaco e la sua disponibilità per i malati sono cose diverse. La scoperta del proteasoma non è arrivata subito, ma è stata un processo graduale. È come creare un fiume da una piccola sorgente, e il fiume è la credibilità scientifica che devi guadagnare agli occhi dei tuoi colleghi: ad esempio si pensava che inibire il proteasoma avesse conseguenze irreparabili, così nessuno credeva alla possibilità di usare il proteasoma come target terapeutico. Inoltre alcune scoperte, come quella alla base del bortezomib, possono arrivare per serendipity e non come risultato di una programmazione. Alla fine, temo che sia fisiologico aspettarsi un periodo di 15-20 anni tra il concept iniziale e la disponibilità del farmaco. Ci sono anche questioni tecnologiche, pensiamo ad esempio al microRNA: lo conosciamo da più di 20 anni, ma non ci sono ancora farmaci microRNA-based. Nel 2006 Fire e Mello hanno vinto il Premio Nobel per la medicina per i loro risultati nel campo della RNA interference: non ci sono ancora applicazioni terapeutiche. E gli esempi potrebbero continuare: oggi si parla molto di CRISPRs, delle loro potenzialità ma anche dei rischi che comportano.
Poi non ci sono solo gli aspetti scientifici, ma anche quelli etici. Possiamo “correggere” i geni? C’è la questione dell’eugenetica: molti vorrebbero figli alti, biondi, intelligenti... C’è molta discussione nella società su questi e altri temi: è un possibile fattore di rallentamento della ricerca scientifica, ma è giusto così. È meglio per la società essere ragionevolmente slow (che non vuol dire impiegare secoli) e avere la sicurezza che la nuova molecola terapeutica colpisca esattamente e selettivamente il target per il quale è stata progettata.
Dall’altra parte ci sono le aspettative dei malati per i quali non esiste una cura efficace, che fanno affidamento nella ricerca scientifica per la disponibilità di nuovi farmaci in tempi brevi.
Senz’altro, ma ci sono stati progressi significativi nella terapia anticancro. Pensiamo ad esempio alle applicazioni della T-cell modulation alla cura del melanoma5. Anche i clinical trial possono richiedere tempi lunghi, nell’ordine di 5 anni o più, e sono necessari per stabilire la reale efficacia dei nuovi farmaci e consentire la loro approvazione: dobbiamo essere sicuri che il nuovo farmaco sia un reale miglioramento rispetto alla situazione preesistente. Alla fine, purtroppo, i veri animali oggetto dei test di laboratorio non sono i conigli o i topi ma siamo noi esseri umani.
L’eterogeneità6 dei profili genetici delle cellule tumorali è uno degli aspetti fondamentali che rendono difficile la lotta contro il cancro...
Sì, in effetti pazienti con un quadro clinico molto simile possono avere landscape genetici totalmente distinti, così da rendere possibili risposte anche molto diverse alla stessa terapia. Inoltre esiste anche l’eterogeneità intratumorale, ad esempio tra le cellule del tumore primario e quelle delle metastasi che interessano la stessa persona. In questi casi gli oncologi adottano terapie basate sull’uso di farmaci diversi, ognuno per attaccare un target specifico.
Dunque l’esistenza di un substrato molecolare comune delle cellule tumorali potrebbe facilitare la ricerca di una cura funzionante su una classe più ampia di patologie. Potrebbe essere l’ubiquitin system questo target comune per lo sviluppo di terapie più generali?
Forse, ma fino a un certo punto. Vediamo ad esempio che gli inibitori del proteasoma funzionano per il mieloma multiplo e per i linfomi non Hodgkin, ma non possiamo dire lo stesso per altri tipi di tumori7. In realtà il termine “cancro” è usato per indicare centinaia di patologie diverse.
Nel 1984 lei è rientrato al Technion (l’Institute of Technology di Haifa) dove, oltre ad un periodo di attività scientifica entusiasmante, ha conosciuto anche qualche problema: il Technion è sostanzialmente una scuola di ingegneri, che spesso percepivano le life sciences come una materia estranea e sono anche arrivati a ventilare la chiusura del vostro laboratorio. Le cose sono cambiate da allora?
Penso di sì, in meglio. Si comincia a capire che la biomedicina e le life sciences non possono fare a meno dell’ingegneria: si pensi alla risonanza magnetica (MRI), alle valvole cardiache, agli organi artificiali. Oggi siamo più consapevoli dell’importanza della multidisciplinarietà: non c’è scelta, gli specialisti devono convivere e collaborare, specialmente per la medicina che ha davanti a sé sfide così complesse. Dunque la situazione è migliorata: non è ancora ottimale [sorride] ma è migliorata rispetto al passato.
Una cosa difficile da comprendere per chi, come fisici e ingegneri, è abituato ad un approccio meccanicistico/deterministico, è che a volte biomarkers molecolari per una determinata patologia si basano su liste di geni completamente diverse tra loro, si vedano ad esempio i tools commerciali prognostici e predittivi per il tumore al seno: sembra quasi che non ci sia un rapporto causa-effetto unico tra genetica e malattia (e risposta alla terapia)...
Pensiamo alla psichiatria, o all’autismo: lì la situazione è ancora più complicata. È un problema generale, legato all’origine multigenica di molte malattie e al diverso contributo dei singoli geni alla patogenesi molecolare. Abbiamo bisogno di sequenziare un gran numero di genomi di pazienti, per comprendere meglio la rete complessa di relazioni tra i diversi geni che contribuiscono alla malattia, e poi cercare di identificare tra di essi pochi geni fondamentali (se esistono) che occupano gli “incroci” piu’ importanti di queste reti [si pensi anche all’importanza crescente attribuita ai gene-pathways8].
C’è una grande discussione su questi temi. Alcuni “guru” della ricerca sul cancro, come Weinberg e Hanahan, suggeriscono di cercare di inquadrare il problema nel suo insieme, con una vista complessiva, piuttosto che analizzare i dettagli fini. Più di un decennio è passato dal primo sequenziamento di genoma umano ma le ricadute terapeutiche sono ancora poche: e ci sono anche gli aspetti economici da considerare, perché gli studi su grande scala basati sulle nuove tecniche di profiling genetico possono essere molto costosi. Ci sono anche questioni di metodologia, ad esempio su quale sia l’approccio più proficuo per la ricerca. Tanti aspetti che rendono la medicina personalizzata dei prossimi anni un settore di ricerca estremamente complesso, ed è per questo motivo che dobbiamo cercare di avere una visione d’insieme del problema, piuttosto che focalizzare i particolari.
Oggi più che mai la medicina fa un largo uso della statistica, e usa termini e concetti a volte difficili da capire per il paziente...
La statistica ha ovviamente un ruolo molto importante, ma deve comunque occupare una posizione di background nel quadro della medicina personalizzata. Se una donna si sente dire dal proprio oncologo che ha un tumore al seno con, diciamo, il 60% di probabilità di guarigione, questo numero deriva dalla formazione culturale dell’oncologo, i suoi studi, la sua esperienza, quello che ha letto nella letteratura scientifica. Ma alla fine il paziente ha una sensibilità, una vita, degli affetti, forse una famiglia, dei figli... è una persona, non un numero. La statistica può permettere di condensare la conoscenza relativa a un gran numero di pazienti oggetto di studi passati per collocare questa paziente in una determinata categoria e capire qual è la terapia più adatta a lei. Oggi è tutto quello che possiamo fare: ma in futuro il medico potrà fare prognosi molto più specifiche e “ritagliate” sul paziente. Non a caso il presidente Obama ha parlato di precisione, aggiungendo un quinta “p” alle caratteristiche della medicina del futuro9.
Sarà una medicina più orientata alla persona e meno alla statistica: vuole dire molto. Sarà un cambiamento profondo.
— F. Maggio
Technical corner
Between 1976 and 1981, Aaron Ciechanover was carrying on his graduate studies with Avram Hershko and Irwin Rose. One of the intuitions that inspired their work was the belief that proteins synthesis and degradation are equally important for cells fitness10.
Disregarding the mainstream attitude of the time, they decided to focus on mechanisms of protein destruction. Contrary to the common belief of single enzyme degradation, they found that several components were called into play: today we believe that the ubiquitin proteasome system (UPS) consists of more than 1000 components. Quite soon they discovered an unusual heat-stable protein involved in protein degradation processes which they called APF-1 (ATP-dependent Proteolysis Factor-1). Under certain conditions, it was capable of surprisingly increasing its molecular weight: it was soon evident that such variation took place when APF-1 attached to substrate molecules in a reversible way (dissociation to substrate could happen as well).
They speculated that covalent attachment of APF-1 was a sort of “tag” with the effect to render the target substrate susceptible to selective degradation by an ATP-dependent protease, followed by the release of free APF-111.
Let's now take a step into the past. In 1975, Gideon Goldstein and colleagues first isolated a small polypeptide hormone, the ubiquitin, apparently present in both prokaryotes and eukaryotes (this is the reason for its name). Later studies revealed that, actually, evidence of presence of ubiquitin in bacteria was a mistake due to laboratory procedure error: still, the name was maintained. More importantly, in 1980, it turned out that APF-1 and ubuquitin were the same protein12.
Some 35 years later, the mechanism by which a polyubiquitinated protein is targeted to the proteasome is not completely clear, yet. We know that coordinated reactions of three different enzymes, E1s, E2s, and E3s, take place, as depicted in Figure 1. Three main steps have been identified13:
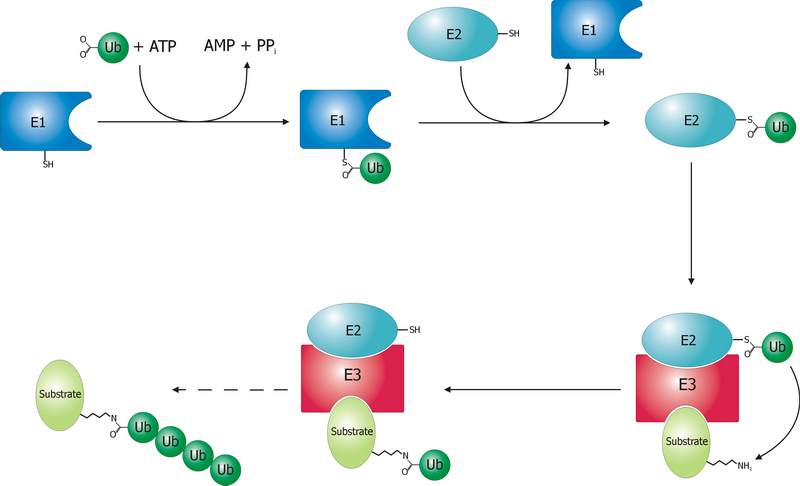
- Activation is a two-step reaction by an E1 enzyme, dependent on ATP, whose results are a ubiquitin-adenylate intermediate and the binding of ubiquitin to a cysteine residue of E1 with release of AMP. In humans two genes, UBA1 and UBA6, are associated to enzymes driving ubiquitin activation.
- Conjugation: the E2 binds to both activated ubiquitin and the E1 enzyme. There are 35 different E2 enzymes in humans, all with highly conserved structure, the UBC (ubiquitin-conjugating catalytic) fold.
- During ligation, the final step, an isopeptide bond between a lysine of the target protein and ubiquitin is created. The target protein is now “tagged” and ready for selective degradation. The human genome is supposed to contain more than 600 E3 ligases.
Protein degradation is then performed by the 26S proteasome, a ubiquitin-dependent large complex of proteins in the cytoplasm (see Figure 2). Substrate proteins tagged with polymeric chain of four ubiquitins or more may bind to 26S: after deubiquitination, proteins are unfolded (by the 19S subcomplex) and then delivered to the 20S proteasome, a second subcomplex where proteolysis occurrs.
Protein degradation by the UPS plays a central role in cell-cycle progression and apoptosis, DNA repair, immune response, cardiac homeostasis, response to cellular stress, et cetera. In addition, involvement of ubiquitin in a number of non-proteolytic processes as biogenesis of organelles and ribosomes, virus infection, regulation of histone modification, has emerged in recent years14.
Not surprisingly, deregulation of the ubiquitin-proteasome system may lead to human pathogenesis. In particular, since the ubiquitin-proteasome system contributes to the proper turnover of both oncoproteins and tumor suppressor proteins, aberrancies in the UPS pathway may result in malignant transformation of cells15. Thus the UPS provides a potential target for anticancer drugs.
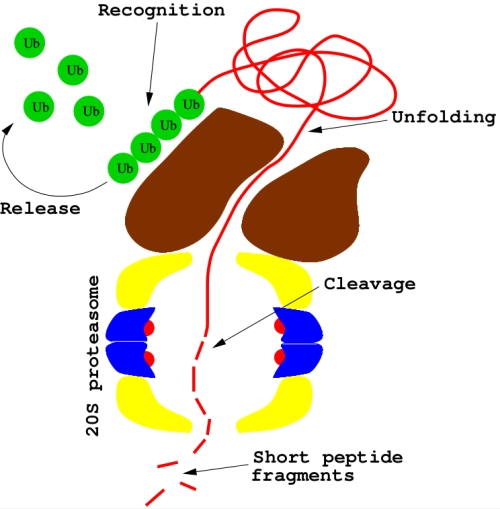
Bortezomib main molecular mechanism is the inhibition of the 20S proteasome (see Figure 2). But why should this be a successful anticancer strategy? This is a difficult question, as many - sometimes competing - molecular processes are called into play16. As a matter of fact, cells with lower proteasome levels are significantly more vulnerable to a proteasome inhibitor, while healthy cells are less sensitive. Proteasome inhibitors may block important tumorigenic pathways, contributing also to sensitize tumors to other anticancer drugs. Furthermore, dysregulation of cell cycle can make cancer cells more susceptible to most proapoptotic stimuli, like p53. The proapoptotic protein NOXA has been recently identified as a key-agent in bortezomib-mediated toxicity in most cancer cell lines. Biological pathways somehow altered by bortezomib are actually numerous and difficult to elucidate: the hypothesis that this drug may activate different mechanisms in different patients has also been proposed.
Bortezomib comes along with two well-known limits.
First, while it is highly effective for hematological cancers, this is not the case for solid tumors. Such behaviour is quite likely explained by the low therapeutic index17 of bortezomib, whose dose-limiting toxicity is associated with peripheral neuropathy. As a consequence, proteasomes abundance in human cells is such that they greatly exceed the number of molecules of the proteasome inhibitor which can be administered to a patient18.
Inherent or acquired resistance is another major problem. In human leukemia THP1 cell-based studies, clones up to 500-fold more resistant to the drug compared with the parental cell line have been found: in that case, the (beta 5) subunit (PSMB5) was overexpressed in resistant cells, resulting in a mutation in the binding site for bortezomib19. In addition, resistance can be the consequence of factors downstream of the proteasome enzymatic complex, like the overexpression of heat shock protein 27 for lymphoma cells.
To overcome these limits, an effort is being made to develop new generation proteasome inhibitors based on mechanisms different from that of bortezomib: most of them are currently in phase I/II clinical trials.
Homage to Alessandro Manzoni (Milan, 7 March 1785 – Milan, 22 May 1873)
Per chi avesse la curiosità di conoscere le ragioni della scelta del titolo, rimandiamo all’estratto del video dell’intervista ↩
L'intervista è stata condotta il 5/6/2015 assieme ad Andrea Mameli all'hotel Panorama di Cagliari, che ringraziamo per l'ospitalità. Un grazie sincero va anche a Saverio Gaeta e agli organizzatori del festival Leggendo Metropolitano che hanno reso possibile questo incontro ↩
A. Ciechanover, Early work on the ubiquitin proteasome system, an interview with Aaron Ciechanover, Cell Death and Differentiation, 12, 1167–1177, 2005 ↩
Il bortezomib come terapia per il mieloma multiplo è stato approvato dalla FDA nel 2003 per uso con i pazienti refrattari, nel 2005 come trattamento di seconda linea, nel 2008 come terapia iniziale ↩
per una review sintetica sull’argomento si veda ad esempio Naidoo J, Page DB, and Wolchok JD, Immune modulation for cancer therapy, BJC, 111, 12:2214-2219, 201 ↩
Gerlinger M, Rowan AJ, Horswell S, Larkin J et al, Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing, NEJM, 366, 10:883-892, 2012 ↩
In particolare per i tumori solidi. Si veda il Technical corner ↩
Vogelstein B, Kinzler KW, Cancer genes and the pathways they control, Nature Medicine, 10, 8:789-799, 2004 ↩
oltre a personalized, predictive, preventive, e participatory: è la “P4 medicine”, espressione molto popolare coniata dal Leroy Hood nel 2005 ↩
Protein degradation is a fundamental task for eukaryotes. Inside cells, proteins homeostasis results from the opposite actions of synthesis and degradation. Proteins typical half-lives vary widely, from minutes to several days: differential rates of protein degradation is a basis of cell regulation. Regulatory molecules, such as transcription factors, are rapidly degraded, as their rapid turnover allows prompt changes to external stimuli. Degradation of other proteins in response to specific signals is another well-known mechanism for regulating the intracellular enzyme activity. Another necessary task is the destruction of faulty or damaged proteins, in order to eliminate the consequences of errors made during protein synthesis. In eukaryotes two major pathways are involved in protein degradation: the ubiquitin-proteasome system and the lysosomal proteolysis. See, for instance, Cooper GM, The Cell: A Molecular Approach. 2nd edition, Sunderland (MA) Sinauer Associates, 2000. ↩
Ciechanover A, Elias S, Heller H, Ferber S, and Hershko A, Characterization of the heat-stable polypeptide of the ATP-dependent proteolytic system from reticulocytes, J. Biol. Chem., 255, 16:7525-7528, 1980 ↩
Wilkinson KD, Urban MK, and Haas AL, Ubiquitin is the ATP-dependent proteolysis factor I of rabbit reticulocytes, J. Biol. Chem., 255, 16:7529-7532, 1980 ↩
Hershko A, and Ciechanover A, The ubiquitin system, Annu. Rev. Biochem., 67:425–79, 1998 ↩
See Nath D and Shadan S, The ubiquitin system, Nature Insight, 458, 7237:421-467, 2009, and references therein ↩
See Table 1 in Micel LN, Tentler JJ, Smith PG, and Eckhardt SG, Role of Ubiquitin Ligases and the Proteasome in Oncogenesis: Novel Targets for Anticancer Therapies, J Clin Oncol, 31, 9:1231-1238, 2013 ↩
For a detailed analysis of molecular mechanisms of bortezomib see Cvek B, and Dvorak Z, The ubiquitin-proteasome system (UPS) and the mechanism of action of bortezomib, Curr Pharm Des, 17:1483-99, 2011 ↩
Roughly speaking, the ratio of the dose of drug that causes adverse effects divided by the dose that leads to the desired pharmacological effect ↩
Dick LR, and Fleming PE, Building on bortezomib: second-generation proteasome inhibitors as anti-cancer therapy, Drug Discovery Today, 15, 5/6:243-249, 2010 ↩
Ruschak AM, Slassi M, Kay LE, and Schimmer AD, Novel Proteasome Inhibitors to Overcome Bortezomib Resistance, JNCI J Natl Cancer Inst, 103, 13:1007-1017, 2011 ↩